 成果展示
软件下载
成果展示
软件下载
简要介绍:
CESSP 是一款基于密度泛函理论(DFT)和动力学平均场理论(DMFT)的量子力学模拟软件包,支持从弱关联材料到强关联材料的电子结构计算、晶体结构与性能预测、分子动力学模拟、热力学物性计算等。
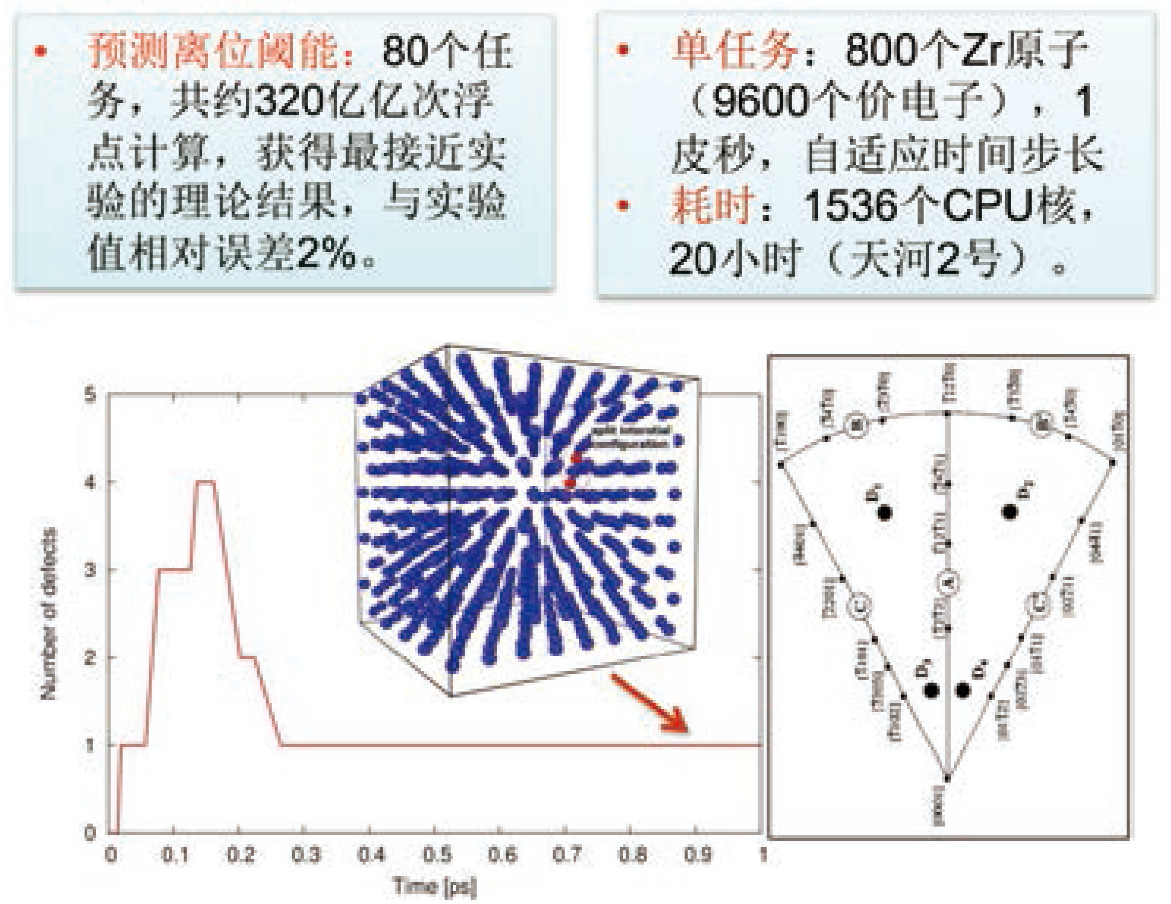
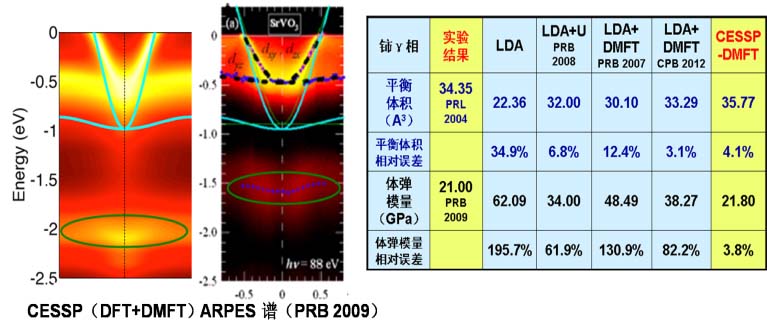
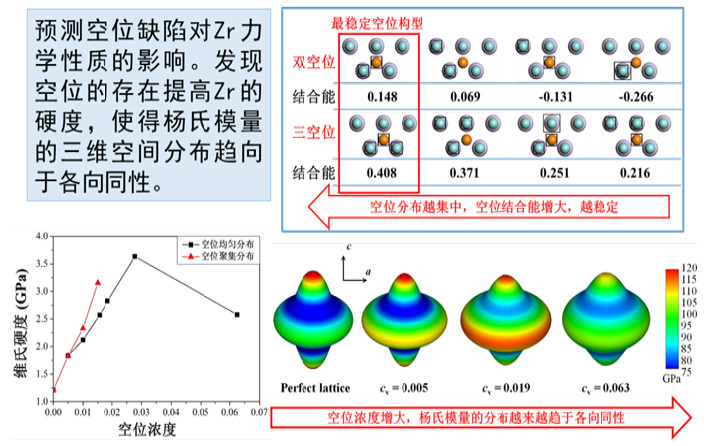
典型示范应用案例:金属材料高压熔点和辐照损伤离位阈能的第一性原理预测,强关联材料SrVO3 电子结构和基态性质计算,含空位缺陷包壳材料锆的力学性质预测等。
主要功能:
1. 电子结构计算和电子强关联效应的修正。
2. 材料结构预测,包括晶体结构和表面重构结构等。
3. 晶体材料力学性质计算。
4. 晶体材料热力学性质计算。
5. 第一性原理分子动力学模拟。
6. 材料电导率和热导率的计算。
技术特色:
1. 基于动力学平均场方法和Gutzwiller变分方法的强关联理论计算;
2. 基于Basin Hopping 算法的晶体结构预测,并通过无缝耦合集成了CALYPSO 软件的功能;
3. 力学性质、热力学性质的快速建模和分析;
4. 系综可配置、时间步长自适应的第一性原理分子动力学模拟,可支持千原子(万价电子)量级体系;
5. 面向高通量的多任务集成计算。
6. 基于Kubo-Greenwood 方法的材料固相、液相的电导率和电子电导率计算。

 请选择版本
请选择版本 请选择类型
请选择类型 请选择类别
请选择类别 请选择操作系统
请选择操作系统 请选择申请
请选择申请